SARS-CoV-2. Di virus “tedeschi” e di virus “padani”
Articolo di Enrico Bucci e Roberto Labanti
Nextstrain è un progetto open source, coordinato da Trevor Bedford (Fred Hutchinson Research Center di Seattle, Washington) che ha lo scopo di favorire la comprensione epidemiologica e di migliorare la risposta alle epidemie grazie, tra l’altro, ad un particolare sistema di visualizzazione dei genomi dei patogeni disponibili pubblicamente. Una particolare sezione del sito, con in questo momento 179 sequenze genetiche depositate nelle banche dati GISAID o Genbank e risultato del lavoro di laboratori di tutto il mondo, è dedicata al virus SARS-CoV-2, responsabile dell’epidemia di COVID-19 in corso. Un articolo dell’ANSA del pomeriggio del 5 marzo, presto ripreso da diversi media, ha suggerito che l’albero filogenetico proposto da Nextstrain indicherebbe che un “focolaio tedesco [di COVID-19] potrebbe avere alimentato silenziosamente la catena di contagi al punto da essere collegato a molti casi in Europa e in Italia”. Ma è proprio così?
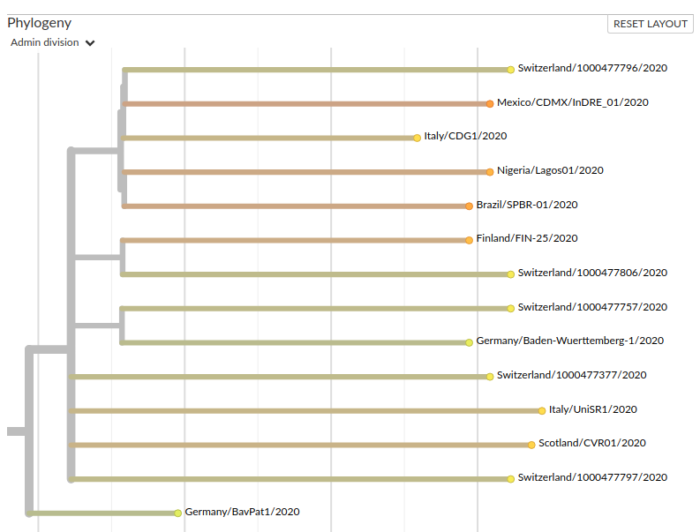
Filogenesi Nextstrain.org del clade A2 alle 16:12 del 06 marzo 2020. E’ è il risultato del lavoro degli scienziati e dei laboratori che hanno raccolto, analizzato e reso disponibili le sequenze genetiche su GISAID e Nextstrain.
Un po’ di ABC
Che cos’è un albero filogenetico? Immaginiamo di tornare indietro nel tempo, supponiamo al momento in cui un singolo virus SARS-CoV-2 ha infettato il primo essere umano. Non appena entrato nel suo involontario ospite, il virus ha cominciato a generare miliardi di copie di sé stesso. Copie molto simili, ma non identiche: ogni copia, in media, contiene infatti circa 30 “errori di copiatura”, vale a dire circa 30 mutazioni nel genoma del virus. Ognuna di queste “copie imperfette”, a sua volta, ha generato miliardi di altre copie ancora meno “fedeli” all’originale, con lo stesso meccanismo.
Capiamo subito che, nel tempo, un virus discendente da un certo “virus antenato” presenterà un certo numero di differenze rispetto al suo progenitore; per accumulo di mutazioni successive ad ogni generazione di virus, cioè, tanto più diverso è un certo virus da un suo progenitore, tante più generazioni sono intercorse.
Confrontando i singoli genomi di due virus, posso stabilire se sono più vicini tra loro – più “imparentati” – o meno, a seconda del numero di mutazioni che li distingue. Inoltre, siccome per produrre una differenza visibile tra due virus ci vuole un certo tempo (perché la maggior parte delle mutazioni casuali dei virus figli da un virus progenitore avranno effetto negativo sui virus, e quindi non le osserverò), posso anche stimare quando si sono differenziati dal loro comune antenato due virus che osservo oggi.
Tracciando su una scala temporale un segno alla data stimata per il loro antenato comune, posso rappresentare quella come la data in cui i due virus che osservo oggi hanno cominciato a differenziarsi, come i rametti di un albero che parta da un nodo comune.
Reiterando la stessa procedura per tutti i virus il cui genoma è stato sequenziato, ottengo un albero come quello in figura.
Il nodo comune ad un certo gruppo di virus rappresenta quindi la data a partire dal quale quei virus si sono differenziati dal loro comune antenato.
Il “focolaio tedesco”
Il 5 marzo i media online hanno rilanciato un articolo (o meglio una letter to the editor) di Rothe et al. apparso sul fascicolo appena uscito della rivista scientifica biomedica The New England Journal of Medicine (NEJM) che descrive un cluster di casi di SARS-CoV-2 (ma qui ancora indicato con il nome provvisorio 2019-nCoV) avvenuto in Germania a partire dal 24 gennaio. Il vettore involontario di questa infezione, che sappiamo avvenuta, da altre fonti, a Stamberg, in Baviera, era stata una partner commerciale di Shanghai che era giunta sul suolo tedesco il 19 e che aveva visitato l’azienda del primo contagiato il 20 e il 21 gennaio, per poi ripartire per la Cina il 22. Per quanto se ne sa, utilizzando anche informazioni successive alla prima pubblicazione della letter sul sito del NEJM il 30 gennaio, il focolaio ha interessato 14 persone (esclusa la cittadina cinese), la cui positività è stata rilevata fra il 27 gennaio e l’11 febbraio.
I tempi della letteratura scientifica non sono però i tempi delle news online e della loro scarsa memoria. Per un cortocircuito informativo, sui media è sembrato che quanto pubblicato fosse una novità e non invece la pubblicazione formale sulla rivista di un qualcosa già presentato alla comunità scientifica e al pubblico a fine gennaio. E, in qualche caso, chi ha letto la “notizia” ha avuto l’impressione che la Germania l’avesse nascosta. Non dovrebbe servire ribadire che non è così.
Su Nextstrain è disponibile, grazie all’Istituto di Virologia della Charite Universitatsmedizin, una sequenza, Germany/BavPat1/2020 (GISAID 406862), ottenuta da un campione raccolto dall’Institut fur Mikrobiologie der Bundeswehr di Monaco di Baviera il 27 gennaio scorso che è, con tutta evidenza, relativa al paziente “1” del cluster bavarese.
Il cluster “padano”
All’interno dell’albero filogenetico, nel raggruppamento di rametti (“clade”) in cui compare Germany/BavPat1/2020 è anche presente un cluster che contiene, ad ora, 13 sequenze, con campioni la cui sequenza genetica è stata determinata in Svizzera (cantone di Zurigo), in Scozia, in Germania meridionale, in Finlandia, in Italia, in Nigeria, in Messico e in Brasile.
Fra queste, le sequenze determinate in Italia sono al momento due. I metadati della prima, Italy/CDG1/2020 (GISAID 412973), dovuta all’Istituto Superiore di Sanità e al Policlinico Militare Celio di Roma e disponibile dal 2 marzo, fanno supporre che il campione, raccolto il 20 febbraio, sia del paziente indice di Codogno (LO), il caso che ha permesso di scoprire che esistevano catene di trasmissione locali del virus. La seconda sequenza, Italy/UniSR1/2020 (GISAID 413489) pubblicata nella notte fra il 5 e il 6 marzo è invece il risultato della milanese Università Vita-Salute del San Raffaele e, ancora dai metadati, è relativa ad una donna 38enne di Milano, con campione raccolto il 3 marzo.
Il 4 marzo, l’Ospedale Sacco di Milano ha annunciato il sequenziamento di altri tre isolati italiani del virus circolante in Lombardia ma al momento in cui scriviamo, queste sequenze non sono ancora disponibili.
Attenzione: il fatto che la sequenza di un virus sia stata determinata in un certo paese, non ci dice che quel virus sia originario di quel paese, visto che, in molti casi, si tratta di isolati in soggetti che provenivano da altre nazioni, e che al loro arrivo nella nazione che poi ha effettuato il sequenziamento genetico sono risultati infetti.
Per questo, come ha riportato l’altro ieri anche uno di noi (EB), le altre sequenze, pur campionate in Europa centrale e settentrionale o, addirittura, in America settentrionale o meridionale (e ora andrà aggiunta quella dell’Africa centro-occidentale) risultano, incrociando i metadati con comunicati stampa istituzionali e notizie pubblicate in questi giorni sui giornali dei rispettivi paesi, essere legate a soggetti provenienti dall’Italia e quindi a trasmissioni avvenute presumibilmente qui da noi, e in particolare, da quello che abbiamo potuto ricostruire finora, a persone transitate per la Lombardia o, comunque, l’Italia del Nord. Possiamo chiamarlo virus “padano” se ciò però non viene inteso come un virus “autoctono”: semplicemente si tratta di un insieme di mutazioni che identifica un gruppo di virus, originati direttamente o indirettamente da un antenato proveniente dall Cina, che ha circolato in Lombardia e qui ha cominciato a mutare, generando una serie di virus simili, che poi sono stati isolati in altri paesi quando alcuni soggetti infetti sono arrivati dalla Lombardia in quei paesi.
I casi “padani” dipendono da quelli tedeschi?
In una serie di tweet del 4 marzo Bedford ha suggerito che, “incredibilmente”, il cluster tedesco contenente Germany/BavPat1/2020 “è il diretto antenato di questi virus più recenti [cioè quelli “padani” NdA] e quindi ha portato direttamente ad una frazione della diffusa epidemia circolante in Europa oggi”. Un’ipotesi che è stata poi ripresa dall’ANSA e quindi da diversi media.
In realtà, per quanto la cosa sia ieri sfuggita in Italia (come racconta anche ValigiaBlu), su Twitter si era immediatamente sviluppata una discussione, un bell’esempio di peer review fuori dagli schemi, con interventi a favore e contro l’ipotesi. Bedford è quindi tornato sulla questione, per specificare che era dispiaciuto e che avrebbe dovuto essere chiaro:
“questa non è una conclusione definitiva. Ulteriori campioni dalla Cina potrebbero mostrare che il caso lombardo è stata un’introduzione distinta. Direi che questa è altamente suggestiva, ma non definitiva”
e ancora, successivamente,
“per chiarire, penso ancora che l’ipotesi della semina bavarese dell’epidemia italiana sia una spiegazione parsimoniosa per [quei] dati genetici, ma ciò non significa che sia l’unica spiegazione”.
Due scenari sono per lo studioso statunitense entrambi plausibili: un passaggio Cina-Germania e poi Germania-Italia, oppure due trasmissioni indipendenti dalla Cina verso le due nazioni. Bedford e collaboratori hanno meglio precisato la questione in un Situation report sempre del 4 marzo circa la somiglianza della sequenza tedesca con il cluster “padano” :
“(sono separati da una sola mutazione), potrebbe indicare una trasmissione non rilevata (“criptica”) in Europa derivante da questo primo cluster tedesco. Potrebbe anche essere il risultato di due introduzioni separate in Europa: una sequenza non campionata da altrove potrebbe posizionarsi tra “BavPat1” e il resto del cluster. Al momento, non possiamo dire con certezza quale scenario sia corretto”.
Possiamo però aggiungere una riflessione. L’albero filogenetico di Nextstrain è costruito anche tenendo conto di una stima del tempo di mutazione. E’ quindi possibile, attraverso questa, ipotizzare una data dedotta (inferred date, ID) massima per la manifestazione delle singole mutazioni.
Orbene: come sappiamo il virus bavarese arriva dalla Cina direttamente, senza alcuna mutazione, con la manager che costituisce il paziente 0 delle infezioni bavaresi, il 19 gennaio; il che piazza l’antenato diretto anche del cluster lombardo in Cina, non in Germania, visto che il cluster lombardo – già mutato rispetto al bavarese – da quel che sappiamo al momento (le stime cambiano con l’aggiunta di nuove sequenze) era già presente in Italia e aveva già iniziato ad irradiarsi con nuove mutazioni quando l’aereo della manager atterra in Baviera.
Pertanto, lo scenario di un’origine bavarese del virus italiano, che tanto appassiona i cercatori di untori e di rivalità nazionalistiche, è molto poco probabile, e le certezze sbandierate su social e sui giornali sono null’altro che ipotesi al momento non supportate e poco probabili.
L’articolo è stato modificato alle 21:40 del 6 marzo per correggere un refuso.
Grazie per il bell’ articolo. Ma ormai penso sia chiaro a tutti che i rimedi hanno fatto danni all’ economia peggiori del male. Faccio una Profezia, che potrete verificare e sbugiardare in prossimità del Capodanno 2021: quando potremo fare bilanci attendibili, la crisi economica scatenata dalla pretesa di fermare un virus influenzale nel 2020 avrà fatto più morti del virus stesso. E non sono complottista, nessuno lo ha pianificato: semplicemente i meccanismi automatici e le linee guida dell’ OMS non considerano la possibilità che convenga accettare qualche centinaio di migliaia di morti in tutto il Mondo (la maggior parte dei quali sono, addirittura, più vecchi di me) piuttosto che bloccare tutto. Quando parte il treno delle 40ene, tutti ci saltano sopra.